
Sanger DNA Sequencing, From Then to Now.
- ClevaLab
- Jun 7, 2023
- 9 min read
Updated: Nov 17, 2024
Explore the basics of Sanger sequencing and the fascinating history behind this groundbreaking technology. This blog post explains the basics of DNA sequencing, including the components necessary for the reaction and the steps involved. Topics also include ddNTPs, cycle sequencing, fluorescent dyes and capillary electrophoresis. Watch the YouTube video or continue on below to find out more.


The 1977 Invention of Sanger Sequencing
In 1977 Frederick Sanger described a method of DNA sequencing using chain-terminating inhibitors. The aim was to determine the sequence of nucleotides in a piece of DNA. This method became known as Sanger Sequencing. These chain-terminating inhibitors are also called ddNTPs.
What Are dNTPs & ddNTPs Anyway?
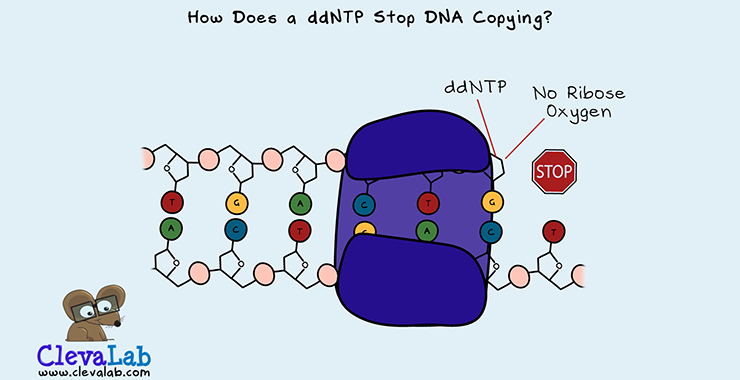
DNA is made up of a chain of 4 different nucleotides called dNTPs. To copy DNA and grow the DNA double strand. DNA polymerase adds the complementary nucleotide. dNTP stands for deoxyribonucleoside triphosphate. A closer look at its structure shows that a dNTP is one deoxyribose (a sugar), a base and a triphosphate. A nucleoside is a ribose sugar and base together. The base is one of the four bases, Guanine (G), Cytosine (C), Thymine (T) or Adenine (A). The sugar is deoxyribose because it has one less oxygen than ribose. ddNTP is short for dideoxyribonucleoside triphosphate. A ddNTP has two oxygens less than ribose, as di- means two.




The role of DNA polymerase is to add new bases to a growing DNA strand. It does this by catalysing a chemical reaction. The incoming dNTP's phosphate group reacts with the bound dNTP's ribose oxygen. This results in the release of two phosphate groups and the addition of the dNTP to the strand. But, if a ddNTP gets added to the strand, there is no ribose oxygen to add another dNTP. This lack of oxygen terminates the DNA chain.



The DNA Naming Convention 5' to 3'
It also makes sense to mention 5' and 3' naming conventions here. 5' and 3' refer to the positions of the carbon atoms in the deoxyribose of dNTP. They're numbered from the carbon linked to the base to the phosphate. The oxygen needed to add new dNTPs to the DNA strand is bound to the 3' carbon. It's common to say that the DNA extends from the 3' end. The other sticky part of the dNTP is the triphosphate. The triphosphate is bound to the 5' carbon. This end of the dNTP is the start, and the 3' end is the finish. When you write down a sequence of DNA, the order of nucleotides is always in the 5' to 3' direction. Also, DNA polymerase only adds a complementary base to a template DNA. So, C pairs with G and A with T.
Steps of the First Sanger Sequencing Method
So how does Sanger sequencing work? The original Sanger Sequencing method is different from the one used today. The original method was manual and used radioactive dyes.
Let's take a look at the original Sanger Sequencing method. We need a primer, DNA polymerase, dNTPs, DNA template and ddNTPs. One of the dNTPs, dATP, is labelled with a radioactive tag. A total of four tubes, one for each ddNTP, are used. The DNA, primer and buffer are heated to 100 degrees. This heat separates the DNA into single strands. Remember, this was before PCR existed. Heating regular DNA polymerase inactivates it. So, it gets added later. Next, the mixture cools to 67 degrees to allow the sequencing primers to bind. Next, we add DNA polymerase, all four dNTPs, and one of the four ddNTPs to each tube. DNA polymerase extends the DNA template. Next, a ddNTP incorporates into the strand, terminating the fragment. The ddNTP is at a lower concentration than the dNTPs, so this incorporation is random. The result is a termination at each base, creating different-length fragments. All fragments in each tube start with the same primer sequence and end in the same nucleotide. Low incorporation of the ddNTP allows the sequencing of longer stretches of DNA. In the original Sanger method, up to 200 nucleotides could be sequenced.











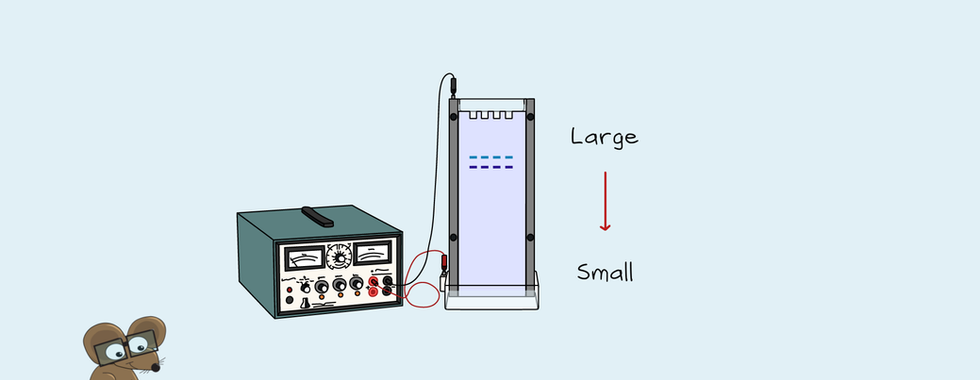


Next, the four sequencing reactions get mixed with a loading dye. Each reaction gets loaded in a separate lane of a polyacrylamide gel. The fragments move through the gel at different speeds depending on their size. The smallest moves the fastest. This type of gel can differentiate a single nucleotide difference in length. At this stage, the fragments can't be seen. The loading dye tells you when the fragments have reached the end of the gel. The sequencing gel gets dried onto a paper support before visualising the fragments. Then, the radiation from the dATPs in the fragments gets detected with X-ray film. This results in bands showing for each fragment. The term used for reading a DNA sequence is "base calling". The DNA is read from 5' to 3' to call the bases. So we start with the shortest fragment first. In this case, it's in the lane of the ddTTP, so the first nucleotide is a "T". The next is in the ddGTP lane and thus is a "G". You continue up the gel based on size to read the whole sequence. So, on this gel, it would read TGCATGCCA.

The First DNA Sequencing Instrument, the AB370A
The original Sanger sequencing method was very labour-intensive. Sequencing 200 nucleotides from only a few samples also took four days. There was a great need to streamline and automate this process. Applied Biosystems created the first commercial sequencing instrument in 1987, the AB370A. Applied Biosystem had already shown that fluorescent dyes could replace radioactive dyes. These are safer and cut out the time needed for X-ray film detection, which took several days. In this instrument, the sequencing reaction had fluorescent sequencing primers. A different coloured fluorescent dye labelled each of the four ddNTP reactions. After sequencing, the four reactions could be mixed and loaded in the same lane of the gel. The AB370A also had a laser that scanned the bottom of the gel. This laser detected the fragments as they passed by. The instrument fed the data into a computer to call the bases automatically. Sixten samples could be run on one gel with a read length of 450 nucleotides.

The AB370A showed that sequencing could be faster and more automated. Scientists started to think sequencing the whole human genome could be within reach. In 1990 the US Government announced the Human Genome Project. This project aimed to map and sequence all the genes in the human genome. By 1990, only <2% of the human genome had been sequenced. Sequencing the human genome would have important implications for science and medicine. It could identify disease-causing and associated genes to treat genetic diseases.
How Does Cycle Sequencing Improve Things?
Another significant improvement in Sanger sequencing happened in the late 80s. Kary Mullis invented PCR in 1983. But, it wasn't until 1989 that Vincent Murray used Taq Polymerase for Sanger Sequencing. In Sanger sequencing, the primer binds to the DNA, and the DNA polymerase extends the fragment. But, as the primer is in excess. Most of the labelled sequencing primers are not extended by DNA polymerase. With Taq polymerase, the DNA can be melted apart after the first extension. Taq polymerase will survive this high heat. The reaction can then be cooled again to anneal another sequencing primer. These melting, annealing and extension cycles repeat the same as in PCR. Many more primers get incorporated into the fragments, increasing the fluorescent signal. But, as there is only one primer, only forward strands and no reverse strands get made. So the same amount of DNA is created each cycle. So it's called linear PCR, later termed Cycle Sequencing. The higher fluorescent signal also meant less DNA was needed for each reaction.




Automated DNA Separation by Capillary Electrophoresis
Another critical advance was in capillary electrophoresis. Capillary electrophoresis is where a small amount of gel is in a fine tube. The DNA is taken in one end, runs through the gel under an electric current and gets detected by a laser at the other end. The fine tube used in capillary electrophoresis allows heat to escape. Therefore, a higher current can be used without the gel overheating. In addition, running the gel under a higher current allows for faster run time and better resolution. Beckman Coulter launched the first commercial capillary electrophoresis instrument in 1989. This launch paved the way for a capillary-based Sanger sequencing system, the ABI PRISM 310. Applied Biosystems launched this system in 1995, and modern Sanger sequencing was born.

ABI PRISM 310, Modern Sanger Sequencing
The ABI PRISM 310 had one capillary for electrophoresis instead of a PAGE gel. One sample could be run in under 3 hours compared to 14 hours with a regular sequencing gel. The sequencing length was also improved and could now sequence up to 600 bp. Preparing a sequencing gel is quite time-consuming and requires skill. With capillary sequencing, gels are no longer needed. The capillary also allowed automation of the sample loading. Up to 96 samples could be loaded in a plate on the system and left to run. Due to electrokinetic injection, low sample volumes and amounts of DNA are needed. This is because DNA is pulled into the capillary by the electric current. The current concentrates it at the end of the capillary. The capillary then moves into a running buffer. Fragments pass through the gel and separate based on size. Then the fragments pass by a laser at the end of the capillary. The size and colour of the fragments get sent to a computer. The software then detects and calls the bases.

Fluorescent ddNTPs and the Invention of BigDyes
While fluorescent ddNTPs were available, sequencing was still performed with fluorescent primers. This is because peak heights were very even with fluorescent primers. Labelled ddNTPs couldn't achieve this even peak height. Not until the introduction of BigDye Terminators in 1997. With fluorescent primers, four reactions are needed. But, with fluorescent ddNTPs, the sequencing reactions can all be in the same tube.

The Instrument that Sequenced the Human Genome
Applied Biosystems continued to improve its system. At the same time, demand continued to grow for the automation of Sanger sequencing. The Human Genome Project needed to make faster progress. By 1998 only 6% of the human genome was sequenced. That same year, Applied Biosystems launched the ABI PRISM 3700 with 96 capillaries. At the same time, they announced a partnership with The Institute of Genome Research (TIGR). TIGR was a not-for-profit institute headed by Craig Venter. Together they formed a new company called Celera and purchased 230 ABI PRISM 3700s. Celera aimed to sequence the human genome faster than the Human Genome Project. It planned to make money by selling access to its sequence data. It also planned to patent genes that could be useful for disease treatment. Profiting from sequencing the human genome was controversial and upset many scientists. The race between public and private sequencing of the human genome had begun.
The ABI PRISM 3700 played a huge role in sequencing the human genome. Each run of 96 samples took less than 2.5 hrs and generated 800 bp of sequence for each sample. With only 15 min of hands-on time by a technician, 1,536 samples could be sequenced daily. With this instrument, the cost per base of sequencing was also reduced. With this new technology, Celera produced a draft sequence of the human genome in three years. Celera published their results in 2001. The Human Genome Project, also aided by the ABI PRISM 3700, published its draft genome at the same time in 2001.

Why Use Sanger Sequencing When There's NGS?
This modified Sanger sequencing method is still used today. But why when there are newer technologies like Next Generation Sequencing? To find out, let's look at how they compare.
Sanger sequencing remains the gold standard for sequencing. It is the method that all other sequencing methods are compared against. Sanger sequencing is the gold standard because it is 99.9% accurate in calling bases. NGS is 99% to 99.9% accurate but depends on the sequencing depth. Sanger sequencing is more cost-effective for sample numbers under 20. It's also faster for this amount of samples. For bigger sample numbers, NGS is more cost-effective and quicker to run. But, the sensitivity of Sanger sequencing to detect a base within a background of other DNA is only 15-20%. Compared to NGS with a 1% sensitivity. Sanger sequencing also has a low sample coverage of one read per sample of only 300-850 bp. In comparison, NGS can generate billions of reads per sample of up to 16 Tb. So big that 128 human genomes can be sequenced in one run. So if you have less than 20 samples or genes you'd like to sequence, Sanger sequencing is still the method of choice.

References:
The original publication describing Sanger Sequencing: Sanger, F., Nicklen, S. & Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74, 5463–5467 (1977).
The use of fluorescent dyes in Sanger sequencing for the first time: Smith, L. M. et al. Fluorescence detection in automated DNA sequence analysis. Nature 321, 674–679 (1986).
The use of fluorescent ddNTPs in Sanger sequencing are described: Prober, J. M. et al. A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science (80), 238, 336–341 (1987).
Automated DNA sequencing and sequencing the Human Genome in 1987: Hood, L. E., Hunkapiller, M. W. & Smith, L. M. Automated DNA sequencing and analysis of the human genome. Genomics 1, 201–212 (1987).
Cycle Sequencing is described for the first time: Murray, V. Improved double-stranded DNA sequencing using the linear polymerase chain reaction. Nucleic Acids Res. 17, 8889 (1989).
The Human Genome Project is announced: Understanding Our Genetic Inheritance, The US Human Genome Project: The First Five Years FY 1991-1995. (US Department of Health and Human Services & US Department of Energy, 1990).
BigDye Terminators were introduced by Applied Biosystems in 1997: Rosenblum, B. B. et al. New dye-labeled terminators for improved DNA sequencing patterns. Nucleic Acids Res. 25, 4500–4504 (1997).
Applied Biosystems and Craig Venter’s TIGR join together to form Celera: Wadman, M. Company aims to beat NIH human genome efforts. Nature 393, 101 (1998).
The ABI PRISM 3700 DNA Analyzer becomes available: PE Biosystems Introduces ABI PRISM 3700 DNA Analyzer for Genomics and Other Large-Scale DNA Analysis Applications. J. Assoc. Lab. Autom. 3, 22–26 (1998).
In 2001 the draft Human Genome is completed by both the International Human Genome Sequencing Consortium and Celera: Craig Venter, J. et al. The Sequence of the Human Genome. Science (80), 291, 1304–1351 (2001) and International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).


